
Disulfide bonds are an integral component of the three-dimensional structure of many proteins. These covalent bonds are found in almost all classes of extracellular peptides and proteins. Disulfides in proteins play an important role in the maintenance of biological activity and conformational stability. After years of accumulation on experience for peptide modification.
Methods to form disulfide bonds
- Air oxidation in aqueous medium. It requires a long duration in basic or neutral pH and a dilution of a peptide. The method is satisfactory for most acidic peptides.
- Thiol-disulfide interchange reaction.
- Powerful oxidizing agents for peptides with a single disulfide bond.
- Mild oxidizing agents for basic and hydrophobic peptides that tend to aggregate and precipitate out of the solutions during the folding process.
Strategy to form two or more pairs of disulfide bonds
- Disulfide bond formation is straightforward in peptides with one pair of cysteine residues. The peptide is simply synthesized via solid or solution phase synthesis, and the solution is then oxidized at pH 8–9.
- Synthesizing peptides that contain two or more pairs of disulfide bonds is more complex. Although disulfide bridges are normally formed toward the end of peptide synthesis, it can be advantageous to couple or elongate chains that include a pre-formed disulfide bond.
- The most widely used groups to protect cysteine residues are Bzl, Acm, tBu, Mob, and TMTr.
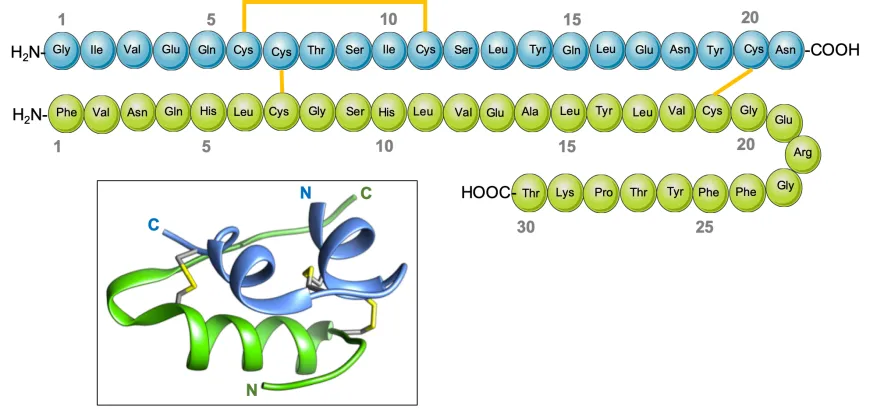
For two or more pairs of disulfide bonds, we will design the cysteine protecting group and disulfide formation sequence first, after the disulfide formation for each pair of disulfide bonds, we will make MS analysis for each disulfide point to identify its correctness and MS analysis report for each disulfide point will be shipped to customers.
